
In parte um deste artigo, revi a estrutura contratual e regulatória aplicada pelo governo dos EUA ao desenvolvimento inicial, fabricação e aquisição das injeções de mRNA da Covid, usando os acordos BioNTech/Pfizer para ilustrar o processo.
Mostrei que a Autorização de Uso Emergencial (EUA) foi concedida a esses produtos com base em ensaios clínicos e processos de fabricação conduzidos com
- sem normas jurídicas vinculativas,
- nenhuma supervisão ou regulamentação de segurança legalmente proscrita, e
- nenhuma reparação legal do fabricante por danos potenciais.
Neste artigo de acompanhamento, fornecerei uma análise detalhada da documentação subjacente.
Outra autoridade/acordo de transação (OTA): um caminho de aquisição militar
A acordo entre o governo dos EUA, representado pelo Departamento de Defesa (DoD), e a Pfizer, representando a parceria BioNTech/Pfizer, em julho de 2020, para a compra de uma “vacina para prevenir a COVID-19” não foi um contrato de aquisição comum.
Foi um acordo sob Outra Autoridade de Transação (OTA) – uma via de aquisição que, de acordo com Diretrizes do Departamento de Defesa, tem sido usado desde 1958 para “permitir que uma agência federal entre em transações que não sejam contratos, doações ou acordos de cooperação. "
[NEGRO ADICIONADO]
Uma revisão completa do uso do OTA pelo DoD, incluindo seu histórico legal, pode ser encontrada no Relatório do Serviço de Pesquisa do Congresso de 22 de fevereiro de 2019. Este relatório, juntamente com todas as outras discussões sobre OTA, especifica que se trata de um caminho alternativo de aquisição para fins de defesa e militares. Não se destina, nem nunca foi utilizado antes da Covid, para qualquer coisa destinada principalmente ao uso civil.
Se você procurar Leis OTA no Código dos EUA, este é o caminho que você seguirá:
Forças Armadas -> Lei Militar Geral -> Aquisição -> Pesquisa e Engenharia -> Acordos -> Autoridade do DoD para realizar determinados projetos de protótipos
Esta via legal mostra muito claramente que as leis OTA se destinam à aquisição de protótipos de investigação e engenharia para as forças armadas.
De acordo com o site da DARPA,
O Departamento de Defesa tem autoridade para três tipos diferentes de TOs: (1) TOs de pesquisa, (2) TOs de protótipo e (3) TOs de produção.
Esses três tipos de TOs representam três estágios de pesquisa inicial, desenvolvimento de um protótipo e eventual produção.
Dentro destes três tipos, existem categorias específicas de projetos aos quais a OTA pode candidatar-se:
- Originalmente, de acordo com o Visão geral da OTA fornecida pelo DoD, a Autoridade de Outras Transações estava “limitada à aplicação a armas ou sistemas de armas propostos para serem adquiridos ou desenvolvidos pelo DoD”.
- OTA foi posteriormente expandido para incluir “qualquer projeto de protótipo diretamente relacionado ao aumento da eficácia da missão do pessoal militar e das plataformas, sistemas, componentes ou materiais de apoio propostos para serem adquiridos ou desenvolvidos pelo DoD, ou à melhoria de plataformas, sistemas, componentes , ou materiais em uso pelas Forças Armadas.”
Até agora, nada disso parece ser uma via de aquisição para milhões de novos produtos médicos destinados principalmente ao uso civil.
Existe alguma exceção para o uso civil de OTA que possa ser aplicada às vacinas de mRNA da Covid?
A Lei de Autorização de Defesa Nacional do ano fiscal de 2004 (PL 108-136) continha uma seção que dava Autoridade para Outras Transações ao “chefe de uma agência executiva que se envolve em pesquisa básica, pesquisa aplicada, pesquisa avançada e projetos de desenvolvimento” que “têm o potencial de facilitar a defesa contra ou a recuperação do terrorismo ou do terrorismo nuclear, biológico, ataque químico ou radiológico.”
Esta disposição foi prorrogada até 2018, mas não parece ter sido prorrogada para além desse ano. Além disso, observe que mesmo neste caso excepcional de uso de OTA não-DoD, a situação deve envolver terrorismo ou um ataque com armas de destruição maciça (QBRN).
Que outras leis OTA podem ser aplicadas?
O relatório CRS de 2019 citado acima fornece este gráfico, mostrando que algumas agências não pertencentes ao DoD têm algumas OTA ou autoridades relacionadas:
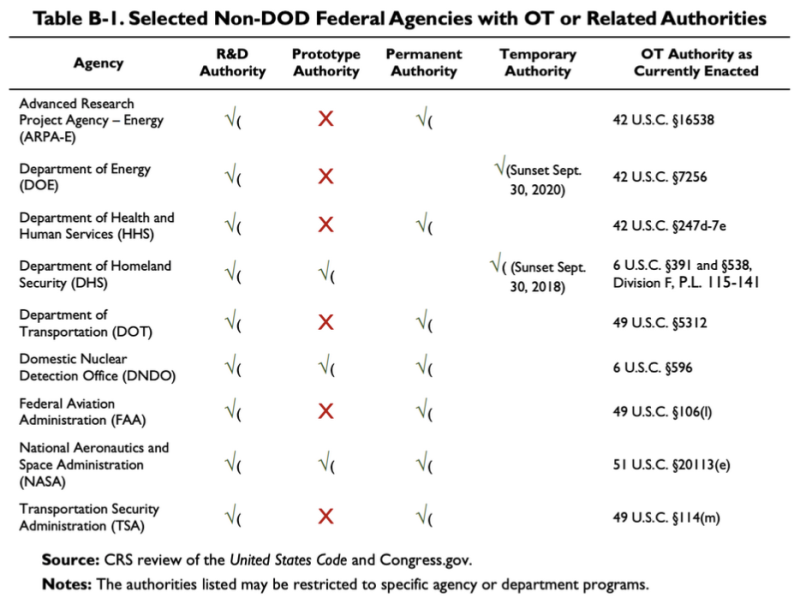
De acordo com esta tabela, o Departamento de Saúde e Serviços Humanos (HHS) possui algumas autoridades de pesquisa e desenvolvimento (P&D) e outras autoridades de transação. A lei relativa ao A Autoridade OT do HHS é 42 U.S.C. §247d-7e.
Onde está esta lei e o que ela diz?
A Saúde Pública e Bem-Estar -> Serviço de Saúde Pública -> Poderes e Deveres Gerais -> Cooperação Federal-Estado -> Autoridade de Pesquisa e Desenvolvimento Biomédico Avançado (BARDA) -> Autoridades de Transação
Portanto, há um lugar na lei relacionada com a saúde e bem-estar civil onde a OTA pode ser aplicável, embora seja válida apenas para pesquisa e desenvolvimento, não para protótipos ou fabricação.
A lei estabelece que o secretário da BARDA tem autoridade do AT
em relação a um produto que é ou pode se tornar um contramedida qualificada ou um produto pandêmico ou epidêmico qualificado, atividades que predominantemente—
(i) sejam conduzidos após pesquisa básica e desenvolvimento pré-clínico do produto; e
(ii) estejam relacionados à fabricação do produto em escala comercial e em um formato que atenda aos requisitos regulatórios do Federal Lei sobre alimentos, medicamentos e cosméticos [21 USC 301 e seguintes] ou abaixo seção 262 deste título.
[NEGRO ADICIONADO]
Os “requisitos regulamentares” enumerados na lei significam que seria impossível para a BARDA/HHS celebrar acordos – mesmo apenas de I&D – para quaisquer produtos médicos (como as vacinas de mRNA) que não fossem submetidos a rigorosos testes de segurança e rigorosa supervisão de fabrico.
“Parceria” do HHS com o DoD contornou as leis de proteção civil
Para resumir a situação de outras autoridades/acordos de transação no que diz respeito às autoridades civis, em geral, e às vacinas de mRNA da Covid, em particular:
- A OTA foi escrita e codificada como uma forma de os militares adquirirem armas e outros sistemas e equipamentos necessários sem muita burocracia. Abrange pesquisa e desenvolvimento, protótipos e fabricação subsequente.
- A única OTA para uma agência de saúde pública é para o HHS e cobre apenas Investigação e Desenvolvimento, não protótipos ou produção.
- Mesmo a OTA de I&D dada ao HHS ainda exige que os produtos sejam fabricados “numa forma que satisfaça os requisitos regulamentares” para a segurança de medicamentos e vacinas.
Por outras palavras: não é possível que o HHS tenha utilizado a sua OTA muito limitada para assinar contratos para centenas de milhões de novos produtos médicos.
Então, o que o HHS fez?
Como observou o Government Accountability Office (GAO) em seu Relatório de julho de 2021 sobre “Contratação Covid-19:” O HHS “fez uma parceria” com o DoD para “alavancar as autoridades OTA do DoD… o que faltava ao HHS”. (P. 24)
Quais são as autoridades de OT do DoD para produtos médicos?
Conforme discutido, o objetivo da OTA é ajudar os militares a obter equipamentos e tecnologia sem muitos problemas burocráticos. Nenhuma das leis originais relativas à OTA mencionou outra coisa senão “plataformas, sistemas, componentes ou materiais” destinados a “aumentar a eficácia da missão do pessoal militar”.
Mas cinco anos antes da Covid, foi introduzido um uso excepcional de OTA:
Em 2015, DoD anunciou o estabelecimento do Consórcio de Contramedidas Médicas CBRN, cujo objetivo era usar o caminho de aquisição da OTA para “trabalhar com o DoD para desenvolver contramedidas médicas químicas, biológicas, radiológicas e nucleares licenciadas pela FDA”. [FDA = Administração de Alimentos e Medicamentos]
Conforme descrito no anúncio de 2015, isto incluía “protótipos de tecnologias para contramedidas médicas terapêuticas visando alvos de toxinas virais, bacterianas e biológicas de interesse para o DoD”. A lista de agentes incluía os principais patógenos da guerra biológica, como o antraz, o ebola e o marburgo.
O anúncio prosseguiu especificando que “as tecnologias facilitadoras podem incluir modelos animais de doenças e patogênese de toxinas virais, bacterianas ou biológicas (múltiplas rotas de exposição), ensaios, tecnologias de diagnóstico ou outras tecnologias de plataforma que podem ser aplicadas ao desenvolvimento de MCMs aprovados ou licenciados [contramedidas médicas].”
Embora isto ainda não se assemelhe à produção de 100 milhões de novas vacinas para uso civil, proporciona mais margem de manobra à OTA do que a muito limitada Autoridade para Outras Transacções concedida ao HHS.
Embora a OTA do HHS exija a adesão a extensas regulamentações de desenvolvimento e fabricação, o caminho da OTA para o DoD desenvolver contramedidas médicas requer apenas “licenciamento da FDA”.
Assim, utilizando outras autoridades de transação do DoD, seria teoricamente possível contornar quaisquer regulamentos de segurança – dependendo dos requisitos para licenciamento da FDA de um produto gerado pela OTA. Como veremos, no caso das vacinas de mRNA contra a Covid, foi concedida Autorização de Uso Emergencial, sem necessidade de qualquer supervisão legal de segurança.
Autorização de uso de emergência (EUA)
Veja como a Food & Drug Administration (FDA) descreve seus poderes EUA:
Seção 564 da Lei FD&C (21 USC 360bbb – 3) permite que a FDA fortaleça as proteções de saúde pública contra agentes biológicos, químicos, nucleares e radiológicos.
Com esta autoridade dos EUA, a FDA pode ajudar a garantir que contramedidas médicas possam ser usadas em emergências para diagnosticar, tratar ou prevenir doenças ou condições graves ou potencialmente fatais causadas por agentes biológicos, químicos, nucleares ou radiológicos quando não houver e alternativas disponíveis (entre outros critérios).
É extremamente importante compreender que estes poderes dos EUA foram concedidos em 2004 em circunstâncias muito específicas relacionadas com a preparação para ataques de armas de destruição maciça, também conhecidas como agentes QBRN (químicos, biológicos, radiológicos, nucleares).
Como explicado no Projeto de Lei de Saúde de Harvard Law,
Em última análise, foi a Guerra ao Terror que daria origem à autorização de uso emergencial. Após os eventos de 11 de setembro de 2001 e os subsequentes ataques com antraz pelo correio, o Congresso promulgou a Lei do Projeto Bioshield de 2004. A lei exigia milhares de milhões de dólares em dotações para a compra de vacinas em preparação para um ataque bioterrorista e para o armazenamento de contramedidas de emergência. Para poder agir rapidamente numa emergência, o Congresso permitiu que a FDA autorizasse produtos formalmente não aprovados para utilização de emergência contra uma ameaça à saúde e segurança públicas (sujeito a uma declaração de emergência por parte do HHS). O registro indica que o Congresso estava focado especificamente na ameaça do bioterrorismo, e não na preparação para uma pandemia que ocorre naturalmente.
A redação da lei dos EUA sublinha o facto de se destinar a ser utilizado em situações que envolvam armas de destruição maciça. Aqui estão as 4 situações em que os EUA podem ser emitidos:
- uma determinação do Secretário de Segurança Interna de que existe uma emergência doméstica, ou um potencial significativo para uma emergência doméstica, envolvendo um risco elevado de ataque com um agente ou agentes biológicos, químicos, radiológicos ou nucleares;
- uma determinação do Secretário de Defesa de que existe uma emergência militar, ou um potencial significativo para uma emergência militar, envolvendo um risco elevado para os Estados Unidos Unidos forças militares, incluindo pessoal que opera sob a autoridade do Título 10 ou Título 50, de ataque com—
- um agente ou agentes biológicos, químicos, radiológicos ou nucleares; ou
- um agente ou agentes que podem causar, ou de outra forma associados a, um risco iminente e específico para a vida dos Estados Unidos Unidos forças militares;
- uma determinação do Secretária que existe uma emergência de saúde pública, ou um potencial significativo para uma emergência de saúde pública, que afecta, ou tem um potencial significativo para afectar, a segurança nacional ou a saúde e segurança dos Estados Unidos Unidos cidadãos residentes no exterior e que envolva agente ou agentes biológicos, químicos, radiológicos ou nucleares, ou doença ou condição que possa ser atribuível a tal agente ou agentes; ou
- a identificação de uma ameaça material de acordo com a seção 319F-2 do Lei do Serviço de Saúde Pública [42 USC. 247d-6b] suficiente para afetar a segurança nacional ou a saúde e segurança dos Estados Unidos Unidos cidadãos residentes no estrangeiro.
Em nenhuma parte destas quatro situações há qualquer menção a uma epidemia natural, pandemia ou qualquer outro tipo de situação de saúde pública que não seja causada por “agentes biológicos, químicos, radiológicos ou nucleares”.
Poderia o SARS-CoV-2 qualificar-se como tal agente?
Se você procurar a definição de “agentes biológicos”No Código Legal dos EUA, você seguirá o seguinte caminho:
Crimes e Processo Penal -> Crimes -> Armas Biológicas -> Definições
Assim, no contexto da legislação dos Estados Unidos, o termo “agentes biológicos” significa armas biológicas, e a utilização de tais agentes/armas é considerada crime.
A Wikipédia fornece isso definição:
Um agente biológico (também chamado de bioagente, agente de ameaça biológica, agente de guerra biológica, arma biológica ou arma biológica) é um bactéria, vírus, protozoário, parasita, fungo, ou toxina que pode ser usada propositalmente como arma em bioterrorismo or Guerra biológica (BW).
Com que base legal foram emitidos EUA para vacinas de mRNA da Covid?
Parece, com base nas leis relativas aos EUA, que nenhuma das quatro situações possíveis descritas na lei poderia ser aplicada a um produto destinado a prevenir ou tratar uma doença causada por um agente patogénico natural.
No entanto, esta lei foi utilizada para autorizar as vacinas mRNA Covid.
Dadas as quatro opções listadas na lei dos EUA, a que foi usada para as “contramedidas” da Covid foi
C) uma determinação do Secretária que existe uma emergência de saúde pública, ou um potencial significativo para uma emergência de saúde pública, que afecta, ou tem um potencial significativo para afectar, a segurança nacional ou a saúde e segurança dos Estados Unidos Unidos cidadãos residentes no exterior e que envolva um agente ou agentes biológicos, químicos, radiológicos ou nucleares, ou uma doença ou condição que possa ser atribuída a tal agente ou agentes.
Quando aplicado especificamente à Covid, foi assim que foi redigido:
o Secretário do Departamento de Saúde e Serviços Humanos (HHS) determinou que existe uma emergência de saúde pública que tem um potencial significativo para afetar a segurança nacional ou a saúde e segurança dos cidadãos dos Estados Unidos que vivem no exterior, e que envolve o vírus que causa o Coronavírus Doença 2019 (COVID-19)…
Não há dúvida aqui de que “o vírus que causa a COVID-19” é considerado equivalente a “um agente ou agentes biológicos, químicos, radiológicos ou nucleares”.
Também é importante notar que a “determinação de uma emergência de saúde pública” da EUA é completamente separada e não depende de qualquer outra declaração de emergência de saúde pública, como as que foram feitas pela OMS, o governo dos EUA , e o Presidente no início da pandemia de Covid-19.
Assim, mesmo quando a OMS, o governo dos EUA e o Presidente declaram que a pandemia acabou, ainda pode haver Autorização de Utilização Emergencial se o Secretário do HHS continuar a afirmar que a situação descrita na secção C) existe.
Olhando todos os EUAs para centenas de produtos médicos relacionados à Covid, é muito difícil ver como o secretário do HHS poderia justificar a afirmação de que “há uma emergência de saúde pública que tem um potencial significativo para afectar a segurança nacional ou a saúde e segurança dos cidadãos dos EUA que vivem no estrangeiro” na maioria, se não em todos, destes casos.
“Critérios legais” adicionais para a FDA conceder autorização de uso emergencial
Uma vez que o secretário do HHS declara que existe uma emergência de saúde pública que justifica a EUA, com base em uma das quatro situações listadas na lei, há mais quatro “critérios legais” que devem ser atendidos para que a FDA emita a EUA . Veja como o FDA explica esses requisitos:
- Doença ou condição grave ou com risco de vida
Para que a FDA emita uma EUA, o(s) agente(s) QBRN referido(s) na declaração de EUA do Secretário do HHS devem ser capazes de causar uma doença ou condição grave ou potencialmente fatal.
NOTA: Este critério repete a especificação de agente QBRN, que é legalmente definido como uma arma utilizada na prática de um crime.
- Evidência de eficácia
Os produtos médicos que podem ser considerados para uma EUA são aqueles que “podem ser eficazes” para prevenir, diagnosticar ou tratar doenças ou condições graves ou potencialmente fatais que podem ser causadas por um(s) agente(s) CBRN identificado(s) na declaração de segurança do Secretário do HHS. emergência ou ameaça de emergência nos termos da seção 564 (b).
O padrão “pode ser eficaz” para EUAs prevê um nível de evidência inferior ao padrão de “eficácia” que a FDA utiliza para aprovações de produtos. A FDA pretende avaliar a eficácia potencial de um possível produto EUA caso a caso, utilizando uma análise de risco-benefício, conforme explicado abaixo.
[NEGRO ADICIONADO]
PERGUNTA JURÍDICA: Como alguém pode alegar legalmente que um produto autorizado sob EUA é “seguro e eficaz” se o padrão legal para EUA é “pode ser eficaz” e a FDA declara que este é um “nível de evidência inferior” do que o padrão usado para aprovações regulares de produtos?
- Análise de risco-benefício
Um produto pode ser considerado para uma EUA se o Comissário determinar que os benefícios conhecidos e potenciais do produto, quando usado para diagnosticar, prevenir ou tratar a doença ou condição identificada, superam os riscos conhecidos e potenciais do produto.
Ao determinar se os benefícios conhecidos e potenciais do produto superam os riscos conhecidos e potenciais, a FDA pretende olhar na totalidade das evidências científicas para fazer uma determinação geral do risco-benefício. Tais evidências, que poderia surgir de diversas fontes, pode incluir (mas não limitado a): resultados de ensaios clínicos nacionais e estrangeiros, dados de eficácia in vivo de modelos animais e dados in vitro, disponível para consideração da FDA. A FDA também avaliará a qualidade e a quantidade do evidência disponível, dado o estado atual do conhecimento científico.
[NEGRO ADICIONADO]
NOTA LEGAL: Não existe uma norma legal e não existem definições legais sobre o que significa que “benefícios conhecidos e potenciais” superam “riscos conhecidos e potenciais”. Também não existe uma definição legal qualitativa ou quantitativa para o que constitui “evidência disponível” aceitável na qual a análise de risco-benefício “pode ser” baseada. Poderia não haver nenhuma evidência real, mas a crença de que um produto tem muitos benefícios potenciais e não muitos riscos potenciais, e isso satisfaria este “requisito legal”.
- Sem alternativas
Para que a FDA emita uma EUA, não deve haver nenhuma alternativa adequada, aprovada e disponível ao produto candidato para diagnosticar, prevenir ou tratar a doença ou condição. Um produto alternativo potencial pode ser considerado “indisponível” se não houver fornecimentos suficientes da alternativa aprovada para satisfazer plenamente a necessidade de emergência.
PERGUNTA JURÍDICA: Além da difamação/proibição flagrante e potencialmente criminosa de tratamentos alternativos para Covid-19, como ivermectina e hidroxicloroquina, em que ponto houve uma alternativa aprovada para “prevenir a Covid-19” (a única coisa para a qual as vacinas de mRNA foram compradas ) – Paxlovid, por exemplo – o que tornaria uma EUA para as vacinas de mRNA não mais legal?
Veja como todos esses “critérios legais” foram satisfeitos na realidade Autorização de uso emergencial para vacinas de mRNA Covid da BioNTEch/Pfizer:
Concluí que o uso emergencial da vacina Pfizer-BioNTech contra a COVID-19 para a prevenção da COVID-19, quando administrada conforme descrito no Escopo da Autorização (Seção II), atende aos critérios para emissão de uma autorização nos termos da Seção 564(c) do a Lei, porque:
- O SARS-CoV-2 pode causar uma doença ou condição grave ou potencialmente fatal, incluindo doenças respiratórias graves, em humanos infectados por este vírus;
- Com base na totalidade das evidências científicas disponíveis para a FDA, é razoável acreditar que a vacina Pfizer-BioNTech contra a COVID-19 pode ser eficaz na prevenção da COVID-19, e que, quando utilizada nas condições descritas nesta autorização, os benefícios conhecidos e potenciais da vacina Pfizer-BioNTech contra a COVID-19 quando usado para prevenir COVID-19 superam seus riscos conhecidos e potenciais; e
- Não há alternativa adequada, aprovada e disponível ao uso emergencial da vacina Pfizer-BioNTech contra COVID-19 para prevenir a COVID-19.
[NEGRO ADICIONADO]
NOTA: O único contexto em que a FDA avaliou os potenciais benefícios e riscos da vacina, e em que a FDA determinou que “pode ser eficaz” foi na prevenção da Covid-19.
Não há qualquer consideração, nenhuma evidência de benefício real ou potencial, e nenhuma determinação de que exista qualquer eficácia potencial para a vacina fazer qualquer outra coisa, incluindo: reduzir o risco de doença grave, reduzir o risco de hospitalização, reduzir o risco de morte , reduzindo o risco de quaisquer condições reais ou potencialmente relacionadas à Covid-19.
PORTANTO, pode-se questionar razoavelmente a legalidade de quaisquer alegações de que a vacina é “segura e eficaz” no contexto de qualquer outra coisa que não seja “quando usada para prevenir a COVID-19” – o que se sabia que as vacinas NÃO FAZEM logo após terem sido introduzido.
Se as pessoas fossem informadas de que as vacinas de mRNA da BioNTech/Pfizer eram “seguras e eficazes” em qualquer outra coisa que não a prevenção da Covid-19, e se fossem ameaçadas com quaisquer consequências por não tomarem a vacina para qualquer outra coisa que não a prevenção da Covid-19, elas poderiam têm um argumento legítimo de que foram ilegalmente coagidos a adquirir um produto não aprovado sob alegações fraudulentas?
Requisitos de terceiro nível para EUA para produtos não aprovados
Assim que tivermos a declaração de emergência específica da EUA, e uma vez que a FDA declare que o produto pode ser eficaz e que qualquer evidência disponível (de zero ao infinito) mostra que os seus benefícios superam os seus riscos (conforme determinado por tudo o que a FDA pensa que esses podem ser), há mais uma camada de regulamentação não relacionada à segurança e à não eficácia.
Aqui está como um Relatório do Serviço de Pesquisa do Congresso de 2018 sobre EUA explica isso:
O FFDCA §564 orienta a FDA a impor certas condições exigidas em um EUA e permite condições discricionárias adicionais quando apropriado. As condições exigidas variam dependendo se a EUA é para um produto não aprovado ou para um uso não aprovado de um produto aprovado. Para um produto não aprovado, as condições de utilização devem:
(1) garantir que os profissionais de saúde que administram o produto recebam as informações necessárias;
(2) garantir que os indivíduos a quem o produto é administrado recebam as informações necessárias;
(3) prever o monitoramento e a notificação de eventos adversos associados ao produto; e
(4) prever a manutenção de registos e relatórios por parte do fabricante.
PERGUNTA LEGAL: Qual é exatamente a “informação necessária?” Sabemos que as pessoas foram informadas de que as vacinas receberam Autorização de Uso Emergencial. Mas foi-lhes dito que isto significa “um nível de evidência inferior” ao exigido para alegações “seguras e eficazes” sobre outros produtos médicos? Foram informados de que existem diferentes níveis de “segurança e eficácia” dependendo se um produto tem EUA ou outro tipo de autorização?
NOTA: A lei exige que haja uma forma de monitorar e relatar eventos adversos. No entanto, não indica quem monitoriza, quais são as normas para a elaboração de relatórios e qual é o limiar para a tomada de medidas com base nos relatórios.
EUA em comparação com qualquer outra via de aprovação de medicamentos/vacinas
Como pesquisador/escritor Sasha Latypova apontou, muitas pessoas ficaram confusas com EUA, porque se parece muito com EAU, que significa “Uso de acesso expandido”. Este é um tipo de autorização concedida a produtos médicos quando há necessidade urgente de um determinado grupo de pacientes (por exemplo, pacientes com câncer em estágio IV cuja expectativa de vida é medida em meses) que estão dispostos a arriscar eventos adversos e até a morte em troca de acesso para um tratamento experimental.
A Autorização de Uso Emergencial não está de forma alguma relacionada nem tem qualquer semelhança com o Uso de Acesso Expandido.
As diversas vias legais para autorizar produtos médicos são apresentadas de forma organizada em uma tabela destacada pelo pesquisador jurídico Katherine Watt. A tabela faz parte de uma apresentação de 2020 para uma Sessão de Aprendizagem Conjunta FDA-CDC: Atualizações regulatórias sobre o uso de contramedidas médicas.
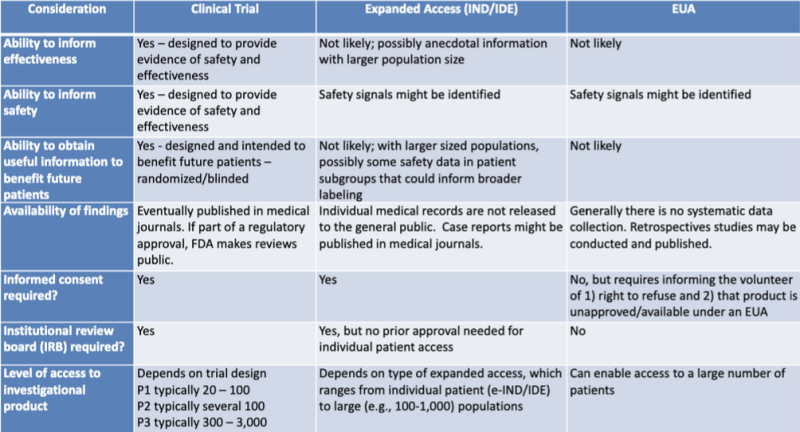
Esta tabela mostra muito claramente que é improvável que o processo de EUA forneça informações sobre a eficácia do produto, não foi concebido para fornecer provas de segurança, não é provável que forneça informações úteis para beneficiar futuros pacientes, não envolve recolha sistemática de dados, não requer estudos retrospetivos, sem consentimento informado e sem conselho de revisão institucional.
Além disso, em um 2009 Instituto de Medicina da publicação Acadêmica Nacional, também destacado por Watt, intitulado “Contramedidas Médicas: Dispensação de Autorização de Uso Emergencial e o Modelo Postal – Resumo do Workshop” encontramos esta afirmação na p. 28:
É importante reconhecer que uma EUA não faz parte do caminho do desenvolvimento; é uma entidade totalmente separada, usada apenas em situações de emergência e não faz parte do processo de aprovação de medicamentos.
Significa isto que as aprovações de contramedidas da Covid-19 baseadas em EUA eram ilegais? Isso significa que não há nenhuma maneira legal de reivindicar que um produto EUA é “seguro e eficaz” porque NÃO FAZ PARTE DO PROCESSO DE APROVAÇÃO DE MEDICAMENTOS?
Conclusão
É eminentemente aparente, dadas todas as informações neste artigo e no anterior Parte 1, que as vacinas BioNTach/Pfizer Covid mRNA foram desenvolvidas, fabricadas e autorizadas ao abrigo de leis militares reservadas para situações de emergência envolvendo guerra biológica/terrorismo, e não para doenças naturais que afectam toda a população civil.
Portanto, a adesão aos regulamentos e à supervisão que esperamos encontrar quando um produto é considerado “seguro e eficaz” para toda a população civil não era legalmente exigida.
Poderá esta análise ser utilizada para contestar a legalidade da afirmação “segura e eficaz” por parte dos funcionários do governo que sabiam o que a EUA implicava? Existem outras ramificações legais?
Eu espero que sim.
É importante ressaltar que nas contestações legais às vacinas de mRNA da Covid apresentadas até agora, não houve decisões (que eu saiba) sobre se a lei militar, como a OTA e a EUA, pode ser aplicada a situações civis. No entanto, houve uma declaração do Juiz do Tribunal Distrital Michael Truncale, no seu demissão do caso do denunciante Brook Jackson v. Ventavia e Pfizer, é importante ter isso em mente.
Aqui, o juiz reconhece que o acordo para as vacinas mRNA da BioNTech/Pfizer era uma OTA militar, mas recusa-se a decidir sobre a sua aplicabilidade às circunstâncias não militares (doença de ocorrência natural, 100 milhões de doses na sua maioria não para uso militar) sob as quais foi emitido:
O facto de tanto militares como civis terem recebido a vacina não indica que a aquisição da vacina fosse irrelevante para aumentar a eficácia da missão militar. Mais importante ainda, a Sra. Jackson está, na verdade, pedindo a este Tribunal que anule a decisão do DoD de exercer outra autoridade de transação para comprar a vacina da Pfizer. Mas, como o Supremo Tribunal dos Estados Unidos há muito sublinha, as “decisões complexas, subtis e profissionais quanto à composição, treino, equipamento e controlo de uma força militar são essencialmente julgamentos militares profissionais”. Gilligan v. Morgan, 413 EUA 1, 10 (1973). Assim, é “difícil conceber uma área de atividade governamental em que os tribunais tenham menos competência”. Eu ia. Este Tribunal não vetará os acórdãos do DoD relativos à eficácia da missão durante uma emergência nacional.
Este é apenas um dos muitos obstáculos legais que permanecem na batalha para, em última instância, proibir todos os produtos de mRNA aprovados durante a emergência da Covid-19, e quaisquer produtos de mRNA subsequentes cuja aprovação tenha sido baseada no processo de aprovação da Covid-19.
Publicado sob um Licença Internacional Creative Commons Attribution 4.0
Para reimpressões, defina o link canônico de volta ao original Instituto Brownstone Artigo e Autor.








