

Nas últimas décadas da minha carreira, passei inúmeras horas trabalhando para proteger os americanos, pesquisando a segurança dos medicamentos. Minha educação e carreira me levaram a passar por cerca de meia dúzia de universidades, pela Big Pharma e pela FDA sob três administrações presidenciais. A segurança de medicamentos considera por que um indivíduo pode tomar um produto farmacêutico e não ter nenhum evento adverso, enquanto um indivíduo diferente pode tomar o mesmo produto, mas ter reações adversas que podem incluir incapacidade permanente ou morte. Por padrão, o estudo da segurança dos medicamentos também considera aspectos não clínicos da fabricação e da qualidade dos medicamentos.
Como a qualidade dos medicamentos é um fator essencial na avaliação da segurança dos medicamentos, minha jornada para proteger os americanos me levou a conceituar e fundar o primeiro “farmácia analítica”Encarregada de verificar cientificamente produtos farmacêuticos de lugares como Índia e China antes de distribuí-los aos pacientes. Infelizmente, a busca pela generosidade em relação à ética e à proteção dos pacientes levou a administração financeira daquela empresa a comprometer-se extenso Violações da FDA e sendo acusado por juízes de fazer falsas alegações científicas (tudo isso ocorreu incoincidentemente após minha saída).
Sem confirmação externa da qualidade do medicamento, os americanos ficam completamente dependentes da FDA e dos fabricantes para avaliar e confirmar a pureza do produto. A segurança dos medicamentos demonstrou ser um problema notável quando se trata de injeções de mRNA da Covid. Infelizmente, se alguém quisesse realizar a sua própria análise sobre injeções de mRNA, não têm uma lista de ingredientes adequadamente detalhada para compará-lo, ou mesmo acesso à metodologia regulatória estabelecida sobre como testá-lo adequadamente quanto à pureza.
É porque os fabricantes e o FDA considera todos os ingredientes dessas injeções de mRNA, incluindo a sequência de propriedades de mRNA mais nanopartículas lipídicas (LNP), incluindo meia-vida, estruturas de LNP, modificação(ões) de superfície, número/tipo(s) de LNPs por dose e pontos de fixação em a fita de mRNA, como não especificada ou “segredo comercial”.
Além disso, o FDA também considera o metodologias sobre como testar a pureza das injeções de mRNA também é um segredo comercial.
Apoio bipartidário e centenas de bilhões de dólares dos contribuintes, mas SEM transparência?
O sigilo do mRNA da Covid existe, embora as administrações Trump e Biden tenham proposto total transparência com as injeções de mRNA, a ponto de suspender os direitos de propriedade intelectual do mRNA da Covid. Apesar disso, tanto a FDA quanto os fabricantes estão permitindo/mantendo controle rígido sobre as patentes, incluindo dados básicos sobre essas injeções, como segredo comercial. Eles estão fazendo isso apesar de todos os fabricantes de vacinas Covid terem recebido centenas de milhões de dólares dos contribuintes de acordo com Forbes/Estatista publicações.
Estudar epidemiologia de segurança de medicamentos já é bastante difícil. Sem pureza/consistência verificável do produto, uma avaliação de segurança completa é impossível.
A total transparência de todos os ingredientes e as medidas de controlo de qualidade são importantes não só porque foram fortemente financiadas pelos contribuintes com centenas de milhões de dólares, mas porque surgiram uma série de questões sobre a segurança e eficácia das injecções de mRNA da Covid.
Além de serem excepcionalmente complexos, sua aprovação foi acelerada pelos reguladores após menos de um ano. A maioria dos medicamentos e vacinas normalmente leva cerca de dez anos para testar totalmente a segurança/eficácia e revisar e aprovar. Além dos ingredientes serem completamente novos, muito complexos e os primeiros do tipo a serem administrados em grande escala, o desenvolvimento incluindo avaliações clínicas de segurança/toxicidade de longo prazo e revisões epidemiológicas foram aceleradas e provavelmente não totalmente elucidadas antes da liberação.
A verificação de ingredientes, transparência e “veracidade” da FDA têm precedentes que datam de 1800:
A verificação analítica e a transparência dos ingredientes ou “verdade na rotulagem” onde o conteúdo da garrafa é requeridos para combinar com os ingredientes listados antecede o estabelecimento do FDA, de volta a 1862. A FDA de hoje nasceu do que começou como um único funcionário do “Departamento de Química” empregado no Departamento de Agricultura dos EUA.
Adulteração, (ingredientes alterados ou tóxicos) marca incorreta (contém um rótulo falso ou é enganoso, ou contém alegações médicas incorretas), ou etiquetagem incorreta (contém ingrediente(s) não listado(s) no rótulo do produto) têm histórias longas e feias na América. Pensava-se que a gravidade tinha atingido o seu pico no início e meados do século XIX – ou pelo menos foi nessa altura que se tornou identificável – já que só em 19 foram desenvolvidos processos técnicos para analisar e detectar fraudes em ingredientes. Antes disso, os chamados “curandeiros viajantes” que se autodenominavam “médicos” (invariavelmente com credenciais duvidosas ou inexistentes) vendiam frascos de produtos “curativos”, cujos rótulos de ingredientes listavam apenas conteúdos nebulosos ou inócuos, como “vitaminas""extratos de ervas,ouóleo de cobra”- ou muitas vezes não tem nenhuma lista de ingredientes.
Naquela época, muitos devotos e puritanos da Nova Inglaterra, que por razões religiosas nunca tocassem em álcool, comprariam essas soluções desses vendedores ambulantes e, sem saber, seriam induzidos a consumir soluções que continham não apenas álcool, mas também narcóticos como ópio e/ou cocaína. Sob o pretexto de melhorar uma cornucópia absurdamente ampla de doenças, os pacientes desenvolveram um vício punitivo e/ou tiveram sua saúde impactada negativamente por esses primeiros “traficantes de drogas”.
À medida que o problema crescia, o governo federal começou a perceber. Eventualmente, o Lei sobre alimentos e medicamentos puros foi aprovada em 1906 e levou à criação da Food and Drug Administration (FDA).
[A FDA tinha um formativo dever de garantir que os medicamentos contenham declarações de rotulagem verdadeiras e atendam a certos padrões de pureza e concentração.
Lembre-se que quase 120 anos exigência de rotulagem verdadeira e parte de “pureza” da Lei de Alimentos e Medicamentos Puros de 1906 enquanto você lê sobre testes de verificação de mRNA e transparência de ingredientes.]
Quais testes de verificação de ingredientes “verdadeiros” e “puros” estão ocorrendo em produtos regulamentados pela FDA?
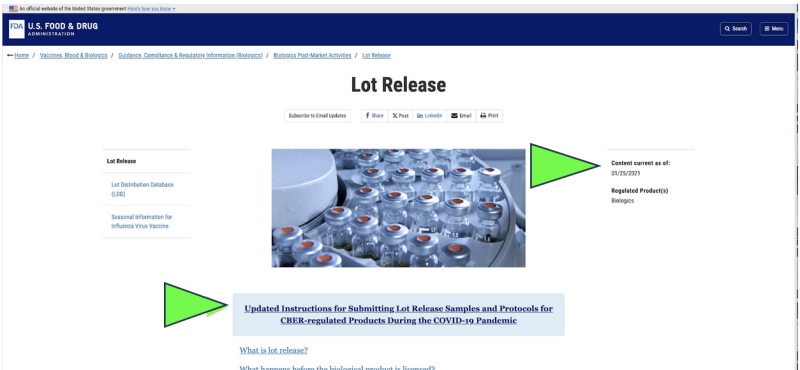
Em 2021, a FDA optou por começar a monitorar a qualidade farmacêutica da América por meio de um coleta remota of envio de amostras por correio para medicamentos em substituição às inspeções de instalações ao vivo por causa da pandemia de Covid. Isso foi legal? Isso poderia ser considerado cientificamente apropriado? Hoje, apesar do fim da pandemia, os únicos testes oficiais de libertação farmacêutica actualmente realizados em qualquer Farmacêutico mRNA Covid aparece para ainda ser feito pelo FDA por meio de um fornecedor fornecido pelo fabricante, “enviado pelo correio” amostra de acordo com um captura de tela do site atual da FDA. Obviamente, um método de amostragem “enviado pelo correio” é muito diferente e potencialmente menos confiável do que a coleta direta de amostras por meio de um método de coleta direta e presencial. Apesar disso, o FDA afirma que “o mais alto padrão em todo o mundo para amostragem e testes. "
Além disso, a FDA está propondo avançar ainda mais em sua política de testes remotos “enviados pelo correio” com um documento de orientação recentemente proposto.
Embora exista apenas como um documento “rascunho” da FDA, os sites oficiais da FDA mostram que o envio de amostras parece já ter sido implementado desde pelo menos janeiro de 2021. A FDA parece estar afirmando os resultados dos testes enviados pelo correio como verificação independente.
Além disso, o parte inferior da primeira página do rascunho da FDA documento propõe expansão de “testes remotos”. Atualmente lista cada Divisão reguladora de produtos da FDA na FDA, o que implica que se trata de uma proposta política para toda a agência.
A lista completa inclui:
- Escritório de Assuntos Regulatórios
- Escritório de Política Alimentar e Resposta
- Escritório de Produtos Combinados
- Centro de Avaliação e Pesquisa Biológica
- Centro de Avaliação e Pesquisa de Medicamentos
- Centro de Dispositivos e Saúde Radiológica
- Centro de Segurança Alimentar e Nutrição Aplicada
- Centro de Produtos de Tabaco
- Centro de Medicina Veterinária
A amostragem de controle de qualidade “enviada” pelo FDA é apropriada? E se as inspeções em restaurantes do Departamento de Saúde dos Estados refletissem a política da FDA?
Essa metodologia de amostragem por “correio” é igualmente absurda, por exemplo, para um departamento de saúde estadual que monitora restaurantes, pedindo-lhes que periodicamente “enviem” vários itens de seu cardápio para uma instalação de teste, para que os departamentos de saúde possam testar possíveis alimentos. contaminação nascida e/ou pedir aos restaurantes que prometam testar eles próprios os itens do menu. E se aquele restaurante fosse na China? E se esse restaurante fosse na Índia? Ou qualquer outro país conhecido por ter um histórico abismal de fraude e controle de qualidade problemas?
Essa metodologia seria inaceitável tanto para restaurantes como para empresas farmacêuticas, por razões que incluem o óbvio: os fabricantes poderiam enviar as amostras que preferirem – não necessariamente amostras representativas de lotes. Obviamente, não é a mesma coisa que os inspetores da FDA adquirem amostras durante inspeções não anunciadas de toda a instalação.
Na analogia do restaurante, é claro que todos os restaurantes enviar amostras de nota “A” o que não seria necessariamente representativo daquilo que os consumidores recebem.

Controle de qualidade: o que são “testes de liberação” farmacêuticos e por que são importantes?
Hoje, o FDA supervisiona a qualidade e o conteúdo dos $2.7 trilhão valor do produto anualmente, mas parece estar suprimindo avaliações e resultados críticos de verificação de ingredientes. A FDA deveria proteger os americanos conduzindo compreensivo testes analíticos como uma soma de verificação para garantir a precisão dos ingredientes. Os resultados disso devem ser transparentes para os contribuintes que financiam o US$ 6.6 bilhões da FDA orçamento. Essa verificação científica é chamada de farmacêutica “teste de lançamento.” Teste de liberação é um termo técnico que se refere a um processo que envolve uma variedade de análises instrumentais usadas para de forma abrangente testar produtos quanto à pureza, concentração, consistência, identidade e impurezas de qualquer tipo.
Todo o FDA nasceu daquele funcionário do “Departamento de Química” de 1862 e da necessidade de transparência e verificação dos ingredientes. Hoje, esse funcionário proliferou em uma todo o departamento da FDA de 1,300 cientistas e pessoal de apoio supostamente dedicado à verificação de ingredientes por meio de testes de liberação farmacêutica. O FDA Escritório de Qualidade Farmacêutica (OPQ) deve garantir que os produtos farmacêuticos correspondam exatamente ao conteúdo dos ingredientes listados, sem variabilidade de qualidade/impureza (qualitativa) ou conteúdo (qualitativa). As regras que exigem isso são muito específicas e detalhadas em 21 CFR § 201.10.
Como o FDA verifica as injeções de mRNA para controle de qualidade:
Os resultados do controle de qualidade dos testes de injeções de mRNA foram particularmente críticos porque são grandes, complexos e foram feitos rapidamente. Embora os contribuintes dependam do FDA para verificar a qualidade da injeção de mRNA e compartilhar os resultados, o FDA parece obrigado a proteger os ingredientes dos fabricantes às custas até mesmo da transparência mais básica em relação aos produtos mRNA Covid. Embora a FDA pareça estar coletando amostras, sua metodologia de “envio por correio” é fundamentalmente falha. Além disso, o FDA não compartilha os resultados desses testes em nenhum lugar onde eu possa localizá-los.
Em outras palavras: durante a pandemia, quando novas injeções de mRNA amplamente implementadas estavam sendo impostas aos americanos em “velocidade extrema” e quando a América confiava mais nos deveres regulatórios/de qualidade do FDA, o FDA estava aceitando “enviados por correio” auto-enviados. in” testes e/ou resultados de controle de qualidade. O FDA não considerou isso Os fabricantes de mRNA admitiram que “lutaram” para responder à fabricação e estavam “lutando” para acompanhar com processos de fabricação? Os fabricantes de ingredientes de mRNA afirmaram ainda que os esforços para atender às necessidades eram “sem precedentes”.
Afirmações como esta não geram confiança na qualidade do consumidor e são ilustrativas da tremenda expansão destes produtos complexos que deveriam garantir especialmente vigilante e exame presencial da FDA sobre instalações e produtos fabricados, pandêmicos ou não. Um fabricante de ingredientes de mRNA, por exemplo, afirmou que repentinamente aumentou sua produção em 50 dobra.
Em meio a essa nova tecnologia promovida em “velocidade extrema”, nenhum dos 1,300 cientistas do OPQ no FDA exigiu inspeções ao vivo, ou pelo menos se ofereceu para fazer outra coisa senão pedir amostras potencialmente questionáveis “enviadas pelo correio” para teste?
A pergunta óbvia é: por que o FDA não coletou amostras diretamente? Mesmo com a pandemia em vigor, o FDA poderia ter inspecionado as instalações usando trajes anti-perigo ou - ou no muito pelo menos – optou por coletar amostras em farmácias, hospitais ou em armazéns distribuidores.
Metodologia oculta para testar ingredientes de injeção de mRNA:
Além da ausência de resultados de testes e de resultados de amostragem questionáveis “enviados pelo correio” – o FDA está adicionalmente ocultar sua metodologia validada, impedindo que outros realizem suas próprias análises independentes sobre a qualidade/pureza das injeções de mRNA.
Analisar de forma independente os medicamentos quanto à pureza e contaminação potencial em comparação com a lista de ingredientes é algo que eu mesmo tentei fazer quando conceituei o primeiro produto do mundo. farmácia analítica. No entanto, uma vez que as injeções de mRNA são uma tecnologia nova com uma lista de ingredientes pouco transparente, a metodologia de teste que seria necessária não é simples como seria para outros medicamentos de moléculas pequenas. Qualquer pessoa que tente pesquisar o armazenamento, a estabilidade, a especificidade, a química, a sensibilidade ou mesmo a metodologia básica para validação e/ou resultados de testes é bloqueada por meio de um relatório da FDA contendo redações ridiculamente invasivas, dificultando até mesmo a compreensão científica mais fundamental de como avaliar potencialmente resultados ou realizar testes impossíveis.
Como exemplo visual comovente, uma única página editada em um resumo regulatório mais longo da FDA (mostrado abaixo) faz parte de um Documento 127-page (das quais apenas 63 páginas foram compartilhadas e, dessas 63 páginas, cerca de 50% foram editadas) sobre como avaliar a pureza, concentração e outras medidas analíticas de injeções de mRNA.
Aqueles Redações da FDA (b)(4) especificou redações detalhadas usadas para “proteger segredos comerciais e informações comerciais ou financeiras confidenciais.” Mas é realmente apropriado rotulá-lo de “comercial” se a pesquisa/desenvolvimento/produto foi financiado com centenas de milhões de dólares dos contribuintes?

Sem uma lista de ingredientes ou metodologia de teste, é impossível para qualquer pessoa fora da FDA ou dos fabricantes saber precisamente como verificar o produto. adulteração (ingredientes alterados ou tóxicos) ou etiquetagem incorreta (porque uma lista completa de ingrediente(s), incluindo a sequência de nucleotídeos e o configurações de nanopartículas lipídicas são particularmente vagas no rótulo do produto).
A falta de metodologia é particularmente problemática, uma vez que novos dados preliminares, utilizando metodologia independente, mostraram evidências de Contaminação de DNA em injeções de mRNA Covid.
Portanto, se um indivíduo externo afirmasse ter testado e encontrado uma impureza em injeções de mRNA e solicitasse a resposta do FDA ou dos fabricantes, ele receberia alguma resposta afirmando algo como:
- Você não usou metodologia de teste validada/apropriada para chegar às suas conclusões e, portanto, suas análises são inválidas.
Para isso, o laboratório independente tentaria solicitar a metodologia de teste a partir da documentação aprovada pela FDA (ou seja, o documento completo contendo Figura 5) perguntando: “Ok, gostaria de testar usando sua metodologia aprovada; você vai nos dizer o que é isso?
- O FDA ou o fabricante responderiam algo como: “O que estamos dispostos a divulgar sobre a metodologia empregada que não seja confidencial pode ser encontrado on-line ou por meio de uma solicitação FOIA da FDA''...onde eles seriam encontrados o seguinte documento fortemente redigido, onde qualquer coisa remotamente significativa é coberta por redações (b)(4).
Lendo nas entrelinhas: é óbvio que tanto os fabricantes quanto o FDA dos Estados Unidos não querem que ninguém além deles conheça os ingredientes completos ou mesmo teste as injeções de mRNA quanto à pureza e consistência.
De acordo com funcionários da FDA: a fabricação farmacêutica é Altamente Propenso a erros:
Muitos coisas podem – e acontecem – dar errado durante o processo de fabricação farmacêutica. Além de possíveis inconsistências com injeções de mRNA/LNP, questões qualitativas e quantitativas implicam cada Produto farmacêutico regulamentado pela FDA. Até mesmo a Câmara e o Senado reconheceram formalmente relatos do fracasso da FDA em proteger a cadeia de abastecimento farmacêutico da América. A maioria dos Farmacêutico da América produto consumidor-usuário finalestá sendo produzido no exterior em países como Índia e China, e outros países com baixos custos de mão-de-obra são não bem conceituado por altos níveis de controle de qualidade. O Registro Federal está repleto de relatos de violações em fábricas indianas e chinesas.
A FDA também está certificando essas plantas – incluindo aquelas com longo histórico de violações – por meio de um sistema de “mail-in” para a FDA? Escandalosamente, a resposta à pergunta é algo que deixaria muito desconfortável qualquer pessoa preocupada com a qualidade farmacêutica.
Enquanto um Seis Sigma O nível de precisão tem sido há muito tempo o objetivo da qualidade e segurança na indústria automobilística, de computadores, de telefones celulares e de outras indústrias de alta tecnologia, mas parece ter sido em grande parte negligenciado quando se trata da fabricação de produtos farmacêuticos.
Funcionários da FDA publicaram dados estimando uma taxa de imprecisão de 2-3σ (sigma) na fabricação de produtos farmacêuticos. Uma qualidade 2σ corresponde a 308,537 defeitos por 1,000,000 oportunidades. (Há provavelmente muito mais de 1,000,000 de oportunidades de erro quando se trata de fabricação farmacêutica.) A FDA está ciente disso nos mais altos níveis de liderança; na verdade, o atual Chefe do Escritório de Qualidade Farmacêutica da FDA, Michael Kopcha até escreveu e publicou o cálculo Six Sigma acima, lamentando a natureza imprecisa da fabricação farmacêutica de volta em 2017.
A latitude de erro para produtos de mRNA e/ou seus LNPs pode ser ainda menos preciso do que 2-3σ, (quanto menor o σ, mais errôneo é o produto), uma vez que incluem material de nucleotídeos e novos LNPs, tornando-os substancialmente mais complexos do que produtos farmacêuticos de moléculas pequenas - apesar de serem desenvolvidos, fabricados e lançados em “ velocidade de dobra.”
Mesmo com o FDA e seus funcionários reconhecendo uma imprecisão inerente à fabricação, por que no vasto mundo dos esportes Será que a FDA não está a cumprir a sua missão de segurança ao partilhar publicamente os seus testes de lançamento da tecnologia mRNA com o público americano que os financia?
Pré-1862 novamente? As injeções de mRNA são as únicas drogas que os americanos não têm Preencha Informações sobre ingredientes?
A falta de clareza sobre o número de sequências de injeções de mRNA e outras informações críticas contrasta diretamente com outro medicamento baseado em RNA aprovado pela FDA – patisiran (Onpattro®). Onpattro fornece de forma transparente a sequência, o peso molecular e a força em miligramas de seus produtos dentro da FDA oficial rotulagem da embalagem conforme ilustrado em um trecho abaixo:


Falta de especificidade da dose de mRNA Covid: 0.3mL (ou 0.5mL) sobre o que?
No momento, ainda não temos informações básicas sobre os ingredientes de qualquer injeção de mRNA da Covid. Os farmacêuticos só sabem dar uma dose específica volume de fluido, e aparentemente o fez sem questionar. Normalmente, a rotulagem oficial da embalagem da FDA deve detalhar os ingredientes reais nesse volume, mas não para os rótulos de mRNA da Covid: eles simplesmente indicam 0.3mL (ou 0.5mL) como “Forma de dosagem e força”.
Além disso, como qualquer estudante do ensino médio poderia lhe dizer, 0.3/0.5mL é um volume, não um força. Não conhecemos quaisquer detalhes quantitativos do que está contido nesses 0.3/0.5mL, tais como: Quantas partículas de LNP? Qual o tamanho/morfologia desses LNPs? Quantas sequências de mRNA nesse volume?




É isso que é considerado adequadamente transparente ou “rotulagem verdadeira” pelo FDA?
O trecho recortado e colado acima da bula contém todas as informações que os fabricantes estão compartilhando com os consumidores em relação à dose - que é lamentavelmente inadequada em comparação com todos os outros rótulos da FDA - ou com qualquer pessoa curiosa para saber algo além da quantidade de fluido para injetar e a concentração de 30 ou 100 mcg de uma sequência de mRNA não especificada.
A notável imprecisão deste rótulo permitida pela FDA parece entrar em conflito especificamente com o seu rótulo de quase 120 anos: “exigir que alimentos e medicamentos contenham declarações verdadeiras nos rótulos e atendam a certos padrões de pureza e concentração. "
É isso que é considerado uma lista “verdadeira” de ingredientes pelo FDA? (Ver 21CFR §352 e 21 CFR §201.10 referente à “declaração de ingredientes” e “medicamentos e dispositivos com marcas incorretas”.)
A questão é: listar ingredientes desconhecidos ou inespecíficos que ninguém, exceto o fabricante, pode decifrar clientes atende ao espírito ou aos requisitos legais de “rotulagem?” Esse rótulo é considerado “verdadeiro” pela FDA americana? De que lado está o FDA, afinal; fabricantes ou consumidores?
Além de não ser diretamente especificado, o número exato de LNP nem de fitas de mRNA em uma injeção de 30 ou 100mcg não pode sequer ser extrapolado estequiometricamente ou com base em Número de Avogadro, porque a sequência de mRNA, o peso molecular e/ou os componentes/configurações do LNP não são fornecidos em nenhum lugar da rotulagem oficial da FDA.
Como alguém pode saber se o número de cadeias de mRNA que codificam a proteína spike para Covid é proporcional à carga de inóculo de Covid que alguém receberia de uma infecção adquirida na comunidade? Responder: eles não podem.
São injeções de mRNA da Covid Rotulado apropriadamente/rotulado incorretamente?
21 CFR 211.125 especifica “Deverá ser exercido controle rigoroso sobre a rotulagem emitida para uso em operações de rotulagem de medicamentos,”Mas parece que o FDA foi muito negligente com a rotulagem aprovada de injeções de mRNA da Covid, apesar do fato de que todos os outros medicamentos – incluindo Onpattro baseado em mRNA – especificam essa informação. Historicamente, as decisões regulatórias da FDA (como quais informações incluir na rotulagem do produto) são baseadas na precedência, e as injeções de mRNA da Covid foram um desvio óbvio da precedência histórica e legal da FDA. Essa notável ausência de dados e falta de clareza remonta aos dias de Cordial de fígado e rim de Morley no final do século XIX. A diferença é: naquela época, o FDA não existia, mas hoje existe um FDA com cerca de 1800 funcionários, pelo menos alguns dos quais acreditavam ostensivamente que este rótulo era transparente e “verdadeiro”.
Declarar um ingrediente desconhecido/indecifrável/obscuro que ninguém jamais poderia determinar com precisão a probabilidade não é o que os legisladores da Pure Food and Drug Act de 1906 pretendiam quando especificaram as regras da FDA sobre “rotulagem verdadeira”. Além disso: o fato de as doses serem duplicadas por volume de diferentes fabricantes (30mcg/0.3mL vs 100mcg/0.5mL) significa que essas sequências de mRNA parecem ser muito diferentes no comprimento dos nucleotídeos e, por sua vez, teriam mais e diferentes LNPs mais anexos. Isso significa que as sequências de mRNA usadas para transcrever a proteína spike têm cerca do dobro do tamanho (10mcg/0.1mL versus 20mcg/0.1mL) em comparação com diferentes fabricantes, ou algo mais está contribuindo para a diferença no comprimento dos nucleotídeos?
Para o leigo que ainda está lendo até agora (parabéns, aliás): A falta de informações detalhadas na rotulagem poderia ser como anunciar amplamente uma casa à venda, afirmando que ela é feita de madeira e tijolos, sobre uma laje de cimento – mas sem mostrar quaisquer fotos da casa (por exemplo, sequência) e não compartilhar sua metragem quadrada (por exemplo, peso molecular). Em qualquer caso, a falta de informação é inadequada e um desvio dos padrões tradicionais.
Todos os outros medicamentos aprovados pela FDA – incluindo outros medicamentos de mRNA – contêm divulgações completas dos ingredientes em seus produtos, incluindo uma representação estrutural e peso molecular de seu produto para que as pessoas saibam exatamente o que estão recebendo.
É verdade: procure qualquer droga que você possa imaginar no Banco de dados Drugs.com e observe como todos os rótulos fornecem estrutura e/ou peso molecular. Prova de que as injeções de mRNA da Covid são um conspícuo exceção à prática histórica de aprovação da FDA e à regra de “rótulo verdadeiro”.
Estudo dinamarquês de 2023 detalha variabilidade clínica significativa entre lotes de injeções de mRNA Covid-19:
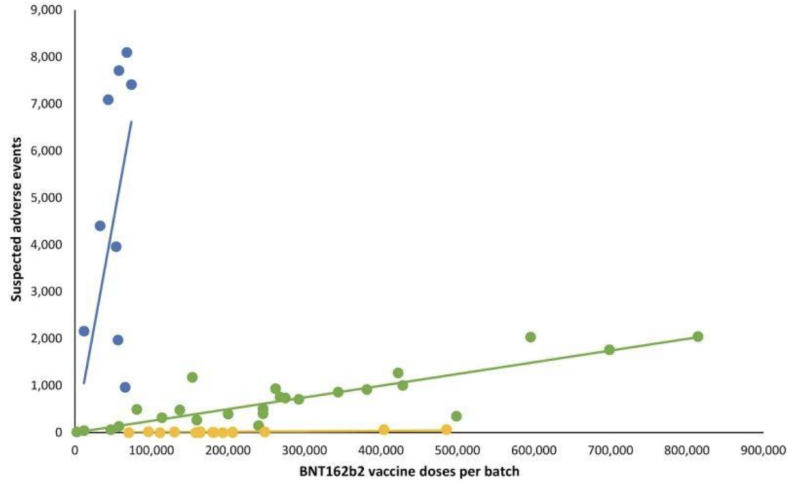
A falta de transparência, mesmo na validação de testes “enviados pelo correio” potencialmente inválidos, parece ter dado aos fabricantes uma aprovação em outra parte extremamente importante do que o FDA supervisiona: possíveis manifestações clínicas em variações de lote/lote de injeções de mRNA. Uma retrospectiva Estudo de segurança dinamarquês publicado no início de 2023 detalhou um padrão altamente desviante de relatos de eventos adversos das injeções de mRNA BNT162b2 da Pfizer-BioNTech, conforme correlacionado com o sistema dinamarquês de notificação de eventos adversos DKMA.
No gráfico de linhas a seguir, diferentes pontos coloridos representam diferentes lotes de injeções de mRNA da Pfizer-BioNTech. Separou os lotes em três categorias diferentes; número alto-baixo a (quase) ausente de grupos de eventos adversos relatados (gráficos azuis, verdes e amarelos, respectivamente).
Em outras palavras: produtos supostamente “equivalentes” do mesmo fabricante parecem ter incidências de eventos adversos muito diferentes, por lote, com cada um desses lotes representando centenas de milhares de injeções de mRNA.
Quando as linhas de regressão linear correspondentes foram adicionadas, surgiu um padrão específico:
Questões importantes sobre a notável disparidade de eventos adversos entre lotes de mRNA da Covid-19 incluem:
- As variações de eventos adversos poderiam ser devidas a variações qualitativas ou quantitativas nas sequências de mRNA ou no número de cadeias de mRNA entre lotes?
- As variações de eventos adversos poderiam ser devidas a variações qualitativas ou quantitativas no tamanho/morfologias ou quantidade de LNPs entre lotes? Quais testes foram realizados para garantir a segurança de vários LNPs usado em injeções de mRNA?
- Esses lotes que correspondiam aos pontos de dados amarelo versus verde versus azul eram de alguma forma qualitativa ou quantitativamente diferentes?
- O armazenamento/manuseio pós-fabricação foi comprometido na instalação de administração (ou em algum outro lugar ao longo da cadeia de abastecimento), levando à variabilidade do produto?
- Qual é a taxa de Sigma/erro deste e de outros produtos provenientes da instalação de fabricação/chefe de turno específico responsável pela fabricação?
- Os ingredientes destes produtos de mRNA da Covid foram provenientes da Índia ou da China ou de outros lugares, dependendo do lote?
- Que porcentagens de lotes de produtos de mRNA da Covid foram testados por meio de coleta pessoal por um inspetor da FDA em comparação com os “enviados pelo correio” desde o início até o momento? Cada lote foi testado usando apenas um desses dois métodos de coleta?
- A FDA realizou verificação de testes de liberação nos lotes do sistema dinamarquês de notificação de eventos adversos DKMA? Se sim, por que o FDA não divulga esses resultados específicos de testes? Se não, por que não foram feitos testes?
- Existe um problema fundamental em produzir consistentemente sequências de LNPs e/ou mRNA de forma confiável e sem contaminação?
Os resultados do estudo dinamarquês e as questões acima sobre eventos adversos poderiam *começar* a ser abordados, mas não sem que a FDA compartilhasse de forma independente os resultados dos resultados dos testes de liberação. Tal como está, devido às redações onipresentes da FDA (b) (4), ninguém conhece a metodologia validada para testar injeções de mRNA da Covid or exatamente quais lotes no estudo dinamarquês foram ou não testados or os resultados desses testes em lote.
…Então, novamente, mesmo que a FDA tivesse optado por divulgar os resultados dos testes em lote, como os consumidores sabem se esses resultados são representativos dos lotes especificados, uma vez que os fabricantes estão auto-selecionando quais amostras “enviar pelo correio”?
Não fornecer transparência aos ingredientes e garantir a qualidade por meio de uma metodologia de amostragem adequada é um requisito fundamental e básico do FDA. Na verdade, foi a principal razão para a formação da FDA! Os americanos não merecem melhor transparência, supervisão e leis de “rotulagem verdadeira” quando se trata dos nossos produtos farmacêuticos – especialmente porque essas leis foram feitas há mais de 100 anos?
Publicado sob um Licença Internacional Creative Commons Attribution 4.0
Para reimpressões, defina o link canônico de volta ao original Instituto Brownstone Artigo e Autor.








